gmx2qmmm
This project is maintained by gmx2qmmm
Welcome to gmx2qmmm
gmx2qmmm is a python interface for Quantum mechanics/Molecular mechanics (QM/MM) calculation.
Overview
gmx2qmmm is a python package to bridge Gaussian and Gromacs. The test runs were performed using Gaussian16 and Gromacs 5.0.2, but the code should be able to read earlier Gaussian and other Gromacs versions. The only limits are the formats of the human-readable input and output files of each program, as such, conversion scripts can be written to make the interface work with any version, if the current code does not support it. Conceptually, gmx2qmmm creates a QM/MM potential and performs either single point calculations (i.e., the current energy of your system), geometry optimizations, and linear relaxed scan. (Other ultilities are ongoing)
System requirements
- python 3.6+
- Gaussian16 (and earlier version)
- Gromacs 5.0.2 (and earlier version)
Downloads
- Download in Git manually
-
git clonegit clone --branch p3 https://github.com/gmx2qmmm/gmx2qmmm_portable.git
gmx2qmmm jobs
| Job type | Calculation |
|---|---|
| Single point calcuation (SP) | Calculate single point energy and forces |
| Geometry optimizations (OPT) | Optimize the system energy via optimizer (Steepest descent, Conjugate gradient or BFGS) |
| Relaxed Scan (SCAN) | Relaxed linear scan (angle and dihedral angle are in development) |
If the calculation is interupted at some point, please check Continue the calculation to continue.
gmx2qmmm input files
usage: gmx2qmmm.py [-h] [-c COORD] [-p TOP] [-n QMATOMS] [-qm QMFILE][-mm MMFILE] [-qmmm QMMMFILE] [-act ACT] [-path PATHFILE] [-g LOGFILE]
| Input files | Command | Default input name |
|---|---|---|
| Coordinate file (.g96 or .gro) | -c | conf.g96 |
| Topology (.top) | -p | topol.top |
| QM atoms file (.ndx) | -n | qmatoms.ndx |
| QM parameters (.dat) | -qm | qm.dat |
| MM parameters (.dat) | -mm | mm.dat |
| QM/MM parameters (.dat) | -qmmm | qmmm.dat |
| Active atoms (.ndx) | -act | act.ndx |
| Path file (.dat) | -path | path.dat |
| Logfile (.log) | -g | logfile |
gmx2qmmm output files
-
Single point calcuation (SP)
File name Description Energy oenergy.txtQM, MM, Link and Total Energy Forces oforces.txtX,Y,Z Forces at each atom -
Geometry optimizations (OPT)
File name Description Energy oenergy.txtQM, MM, Link and Total Energy in each step Forces oforces.txtX,Y,Z Forces at each atom in each step - Set
print_level=NORMALin-qmmmfile : Remain the last step information only - Set
print_level=FULLin-qmmmfile : Remain all information
- Set
-
Relaxed Scan (SCAN)
File name Description Energy oenergy.txtQM, MM, Link and Total Energy in each scan step Forces oforces.txtX,Y,Z Forces at each atom in each scan step Energy oenergy_scanRa-b_step.txtprint_level=FULLin-qmmmfile is required. QM, MM, Link and Total Energy every OPT step in each scan step. a,b:scan atomForces oforces_scanRa-b_step.txtprint_level=FULLin-qmmmfile is required. X,Y,Z Forces at each atom every OPT step in each scan step. a,b:scan atomSince there are many output files in the scan job, the output files are store in a subdirectory of the base directory.
- Example: Linear scan atom1 and atom2 by 3 steps with
print_level=NORMALbase_directory/ |-- scanR/ |--R1-2/ (contains last OPT output) |-- oenergy.txt (Scan energies) |-- oforces.txt (Scan Forces) ... - Example: Linear scan atom1 and atom2 by 3 steps with
print_level=FULLbase_directory/ |-- scanR/ |--R1-2/ (contains all OPT output) |-- oenergy.txt (Scan energies) |-- oforces.txt (Scan Forces) |-- oenergy_scanR1-2_1.txt (OPT energies in scan step 1) |-- oenergy_scanR1-2_2.txt (OPT energies in scan step 2) |-- oenergy_scanR1-2_3.txt (OPT energies in scan step 3) |-- oforces_scanR1-2_1.txt (OPT forces in scan step 1) |-- oforces_scanR1-2_2.txt (OPT forces in scan step 2) |-- oforces_scanR1-2_3.txt (OPT forces in scan step 3) ...
- Example: Linear scan atom1 and atom2 by 3 steps with
Startup examples : glycine serine
The turtorial contains SP and OPT calculation of glycine serine (GLYSER).
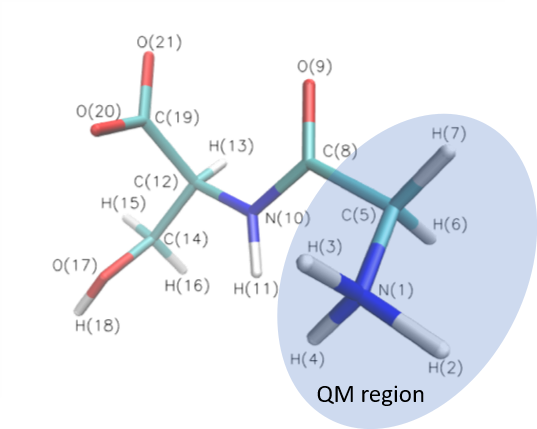
References
A user‐friendly, Python‐based quantum mechanics/Gromacs interface: gmx2qmmm Jan P. Götze, Yuan‐Wei Pi, Simon Petry, Fabian Langkabel, Jan Felix Witte, Oliver Lemke https://doi.org/10.1002/qua.26486
Support and development
For bug reports/suggestions/complaints please raise an issue on GitHub.
Or contact us directly: gmx2qmmm@gmail.com