gmx2qmmm
This project is maintained by gmx2qmmm
Startup example : glycine serine (GLYSER)
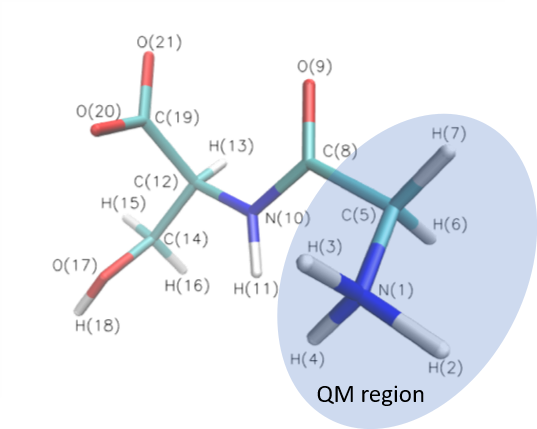
Glycine serine (GLYSER) includes 21 atoms. In this case, we select the first 7 atoms in the QM region and other atoms are in MM region. The figure is showed below.
Examples in example/sp and example/opt
Before running the calculation, please setup paths of the Gromacs and Gaussian
- Single point calculation
- Go to
/example/spdirectory - Run
python ../../gmx2qmmm.py
- Go to
- Geometry optimization
- Go to
/example/optdirectory - Run
python ../../gmx2qmmm.py
- Go to
- Relaxed scan optimization
- Go to
/example/scandirectory - Run
python ../../gmx2qmmm.py
- Go to
Processes from the beginning and customize names of input files
- Prepare Coordinate file (.g96 or .gro) and Topology (.top) files of GLYSER.
- Create QM atom file (.ndx): Since we select the first 7 atoms in the QM region, set QM atom file (.ndx)
1 2 3 4 5 6 7 - Create QM parameters (.dat): The total chages of QM region is 1, so set the parameter charge=1 in QM parameters (.dat). Others are maintained as default.
charge=1 - Create MM parameters (.dat): We use default setting in MM parameters (.dat), so keep it empty.
- Create QM/MM parameters (.dat): If we run
- a SP calculation (default setting), keep QM/MM parameters (.dat) empty.
- a OPT calculation, set parameter jobtype=OPT in QM/MM parameters (.dat)
jobtype=OPT - a Linear SCAN calculation, set parameter jobtype=SCAN in QM/MM parameters (.dat)
jobtype=SCAN- Create a scan file
scan.txt, for example: Linear scan atom1 and atom2 by 3 steps with 0.1 stepsizeR 1 2 0.1 3
- Create a scan file
- Create Path file (.dat): Setup your software path in Path file (.dat)
- Create Active atoms (.ndx): Select active atoms in the calculation, in this case we select all. So set Active atoms (.ndx),
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 - Run
python [path of gmx2qmmm.py ][-h] [-c COORD] [-p TOP] [-n QMATOMS] [-qm QMFILE] [-mm MMFILE] [-qmmm QMMMFILE] [-act ACT] [-path PATHFILE] [-g LOGFILE]